Biopython – 序列输入/输出
Biopython 有一个内置的Bio.SeqIO模块,它提供了分别从文件读取和写入序列的功能。 Bio.SeqIO支持几乎所有生物信息学中使用的文件处理格式。 Biopython 严格遵循单一方法用SeqRecord对象向用户表示解析的数据序列。
序列记录
Bio.SeqRecord模块提供的SeqRecord对象包含序列的元数据以及有关序列的信息。下面列出了一些主要数据信息:
| Record | Description |
|---|---|
| seq | An actual sequence to be parsed. |
| id | Primary identity of the sequence, by default it is string type |
| name | The name of the sequence, by default it is string type. |
| description | Displays the information about the sequence in human-readable format. |
| annotations | Dictionary containing additional information related to the sequence. |
阅读顺序:
Biopython Seq模块有一个内置的read()方法,该方法获取一个序列文件,并根据文件格式将其转换为单个SeqRecord 。它能够解析只有一条记录的序列文件,如果文件没有记录或多于一条记录,则会引发异常。 read()方法的语法和参数如下:
Bio.SeqIO.read(handle, format, alphabet=None)
| Arguments | Description |
|---|---|
| handle | Handle to file or takes filename as string(older versions only take handle) |
| format | File; format as a string |
| alphabet | Optional parameter, used when sequence type is not automatically inferred from file(ex. format = “fasta”). |
Python3
# Import libraries
from Bio import SeqIO
# Reading file
record = SeqIO.read("sequence.gb", "genbank")
# Showing records
print("ID: %s" % record.id)
print("Sequence length: %i" % len(record))
print("Sequence description: %s" % record.description)Python3
# Import libraries
from Bio import SeqIO
# Parsing file
filename = "sequence.fasta"
for record in SeqIO.parse(filename, "fasta"):
# Showing records
print("ID: %s" % record.id)
print("Sequence length: %i" % len(record))
print("Sequence description: %s" % record.description)Python3
# Import libraries
from Bio import SeqIO
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
rec1 = SeqRecord(Seq("MMYQQGCFAGGTVLRLAKDLAENNRGARVLVVCSEITAVTFRGPSETHLDSMVGQALFGD"
+ "GAGAVIVGSDPDLSVERPLYELVWTGATLLPDSEGAIDGHLREVGLTFHLLKDVPGLISK"
+ "NIEKSLKEAFTPLGISDWNSTFWIAHPGGPAILDQVEAKLGLKEEKMRATREVLSEYGNM"),
id="gi|14150838|gb|AAK54648.1|AF376133_1",
description="chalcone synthase [Cucumis sativus]")
rec2 = SeqRecord(Seq("MVTVEEFRRAQCAEGPATVMAIGTATPSNCVDQSTYPDYYFRITNSEHKVELKEKFKRMC"
+ "EKSMIKKRYMHLTEEILKENPNICAYMAPSLDARQDIVVVEVPKLGKEAAQKAIKEWGQP"
+ "KSKITHLVFCTTSGVDMPGCDYQLTKLLGLRPSVKRFMMYQQGCFAGGTVLRMAKDLAEN"
+ "NKGARVLVVCSEITAVTFRGPNDTHLDSLVGQALFGDGAAAVIIGSDPIPEVERPLFELV"
+ "SAAQTLLPDSEGAIDGHLREVGLTFHLLKDVPGLISKNIEKSLVEAFQPLGISDWNSLFW"
+ "IAHPGGPAILDQVELKLGLKQEKLKATRKVLSNYGNMSSACVLFILDEMRKASAKEGLGT"
+ "TGEGLEWGVLFGFGPGLTVETVVLHSVAT"),
id="gi|13925890|gb|AAK49457.1|",
description="chalcone synthase [Nicotiana tabacum]")
sequences = [rec1, rec2]
# Writing to file
with open("example.fasta", "w") as output_handle:
SeqIO.write(sequences, output_handle, "fasta")
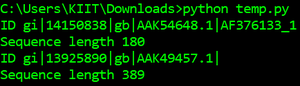
for record in SeqIO.parse("example.fasta", "fasta"):
print("ID %s" % record.id)
print("Sequence length %i" % len(record))输出:
诵经顺序:
当我们必须从句柄中读取多条记录时,使用Bio.Seq模块提供的Parse()方法。它基本上将序列文件转换为返回SeqRecords的迭代器。如果文件包含字符串数据,则必须将其转换为句柄来解析它。无法确定字母表的文件格式,明确指定字母表(例如 FASTA)很有用。 parse()方法的语法和参数如下:
Bio.SeqIO.parse(handle, format, alphabet=None)
| Arguments | Description |
|---|---|
| handle | Handle to file or takes filename as string(older versions only take handle) |
| format | File format as a string |
| alphabet | The optional parameter, used when sequence type is not automatically inferred from file(ex. format = “fasta”). |
蟒蛇3
# Import libraries
from Bio import SeqIO
# Parsing file
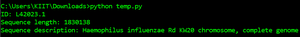
filename = "sequence.fasta"
for record in SeqIO.parse(filename, "fasta"):
# Showing records
print("ID: %s" % record.id)
print("Sequence length: %i" % len(record))
print("Sequence description: %s" % record.description)
输出 :
写入序列:
为了写入文件Bio.Seq模块有一个write()方法,它将序列集写入文件并返回一个表示写入记录数的整数。确保在调用句柄后关闭句柄,否则数据会刷新到磁盘。 write()方法的语法和参数如下:
Bio.SeqIO.write(sequences, handle, format)
| Arguments | Description |
|---|---|
| sequences | List or iterator of SeqRecord object(or single SeqRecord in Biopython version 1.54 or later) |
| handle | Handle to file or takes filename as string(older versions only take handle) |
| format | File format to write as a lowercase string |
注意:要下载文件,请单击此处
蟒蛇3
# Import libraries
from Bio import SeqIO
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
rec1 = SeqRecord(Seq("MMYQQGCFAGGTVLRLAKDLAENNRGARVLVVCSEITAVTFRGPSETHLDSMVGQALFGD"
+ "GAGAVIVGSDPDLSVERPLYELVWTGATLLPDSEGAIDGHLREVGLTFHLLKDVPGLISK"
+ "NIEKSLKEAFTPLGISDWNSTFWIAHPGGPAILDQVEAKLGLKEEKMRATREVLSEYGNM"),
id="gi|14150838|gb|AAK54648.1|AF376133_1",
description="chalcone synthase [Cucumis sativus]")
rec2 = SeqRecord(Seq("MVTVEEFRRAQCAEGPATVMAIGTATPSNCVDQSTYPDYYFRITNSEHKVELKEKFKRMC"
+ "EKSMIKKRYMHLTEEILKENPNICAYMAPSLDARQDIVVVEVPKLGKEAAQKAIKEWGQP"
+ "KSKITHLVFCTTSGVDMPGCDYQLTKLLGLRPSVKRFMMYQQGCFAGGTVLRMAKDLAEN"
+ "NKGARVLVVCSEITAVTFRGPNDTHLDSLVGQALFGDGAAAVIIGSDPIPEVERPLFELV"
+ "SAAQTLLPDSEGAIDGHLREVGLTFHLLKDVPGLISKNIEKSLVEAFQPLGISDWNSLFW"
+ "IAHPGGPAILDQVELKLGLKQEKLKATRKVLSNYGNMSSACVLFILDEMRKASAKEGLGT"
+ "TGEGLEWGVLFGFGPGLTVETVVLHSVAT"),
id="gi|13925890|gb|AAK49457.1|",
description="chalcone synthase [Nicotiana tabacum]")
sequences = [rec1, rec2]
# Writing to file
with open("example.fasta", "w") as output_handle:
SeqIO.write(sequences, output_handle, "fasta")
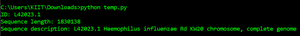
for record in SeqIO.parse("example.fasta", "fasta"):
print("ID %s" % record.id)
print("Sequence length %i" % len(record))
输出: